
Trisomie 21
Atlas of Genetics and Cytogenetics in Oncology and Haematology
TRISOMIE 21
Maladie chromosomique viable la plus fréquente.
Comme toutes les autres maladies chromosomiques autosomiques, associe dysmorphie + retard psychomoteur: rechercher les malformations associées, fréquentes (>1/3 des cas): établir une prise en charge medico-pédagogique.
I. EPIDEMIOLOGIE
(Une question sur l'épidémiologie fera envisager les risques de récurrence en fonction du caryotype - voir le paragraphe caryotype)
1,5 /1 000 naissances.
sex ratio: 3 garçons / 2 filles.
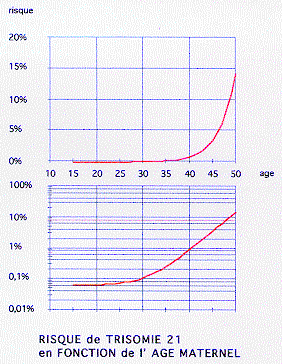
âge maternel moyen augmenté (34 ans).
fréquences maximales: à 28 ans (mais ceci correspond au pic maximal de la natalité)
et vers 37 ans en effet, le risque augmente avec l'âge maternel: <0,1% au dessous de 30 ans; entre 0,1% et 1% entre 30 et 40 ans (0,2% à 34 ans, 0,5% à 38 ans, 0,7% à 39 ans); >1% au dessus de 40 ans (5% à 46 ans, 15% à 50 ans).
II. EXAMEN CLINIQUE
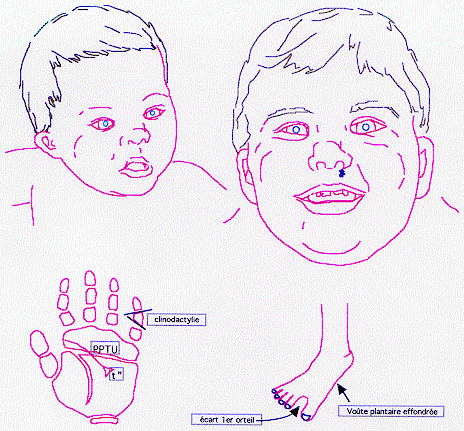
1 - Syndrome dysmorphique, associant à divers degrés:
visage évocateur +++:
microcéphalie fréquente; cou court; nuque plate;
faciès lunaire;
angle naso frontal effacé;
aspect des narines en "prise de courant";
hypertélorisme ou pseudo hypertélorisme;
épicanthus (régresse avec l'âge);
fentes palpébrales en haut et en dehors;
taches de Brushfield dans l'iris (pathognomonique, visible sur yeux bleus).
macroglossie; glossite exfoliatrice; langue scrotale (chez l'adolescent et l'adulte);
bouche souvent ouverte;
palais ogival, étroit;
retard de dentition (anomalies de nombres, agénésie des incisives latérales...);
mains et pieds:
courts et trapus;
brachymésophalangie du 2e et du 5e doigts;
clinodactylie du 5e;
voute plantaire effondrée;
ler orteil séparé des autres par un sillon.
peau sèche, marbrée (livedo), infectée au pourtour des orifices.
hyperlaxité ligamentaire.
diastasis des droits; hernie ombilicale fréquente.
2 - Retard psychomoteur, constant:
hypotonie +++ dés la naissance (tenue de tête à 6 mois, tenue assise à 1 an, marche à 2 ans).
le retard mental, peu net chez le nourrisson, s'établit rapidement.
enfants:
affectueux, doux, joyeux;
difficultés de langage;
aiment jouer, mimer, ranger tous les objets; enfants minutieux;
mémoire normale.
épilepsie possible (chez 3% à 9%, et dans 1% de la population générale).
3 - Dermatoglyphes:
- pli palmaire transverse unique (pptu) dans 75 % des cas; mais le pptu est aussi présent dans 1 % de la population non trisomique; par conséquent, sur 8 pptu vus à la naissance, 7 appartiennent à la population générale et seulement 1 à un bébé trisomique 21 (risque de 1% X 699/700 naissances versus risque de 75% X 1/700 naissances): un pptu isolé NE DOIT PAS faire porter le diagnostic de trisomie 21, diagnostic qui risquerait d'entrainer un choc psychologique profond, voire rémanent, chez les parents. - triradius axial en t''(75 % des cas).
- indice de transversalité (it) > 30.
Cette association de signes impose la recherche de malformations qui peuvent grever le pronostic vital et nécessiter une thérapeutique urgente.
4 - Malformations (45% des cas):
Cardiaques (40 %):
canal atrio-ventriculaire (10 %);
communication inter ventriculaire (CIV) (10 %);
communication inter auriculaire (CIA) (5 %);
persistance du canal artériel (CA) (5 %)...
Digestives ( 10 %):
sténose duodénale (1/3 des sténoses duodénales se trouvent chez les trisomiques 21);
imperforation anale...
Oculaires:
cataracte (congénitale ou acquise);
astigmatisme;
myopie;
strabisme;
glaucome congénital;
nystagmus.
5 - Autres:
Hématologiques: parfois leucoblastose transitoire pouvant rechuter en leucémie aiguë lymphoblastiques (LAL) on non lymphoblastiques (LANL) plus souvent; les leucémies mégacaryocytaires (LANL M7) sont particulièrement fréquentes; attention à l'hypersensibilité au methotrexate.
Immunologiques: hyporéactivité à la tuberculine; déficit immunitaire.
Métaboliques: hyperuricémie; glycémie perturbée; TSH souvent augmentée; hypo ou hyperthyroïdie.
III. DIAGNOSTIC: LE CARYOTYPE
confirme le diagnostic, permet le conseil génétique:
le risque de récurrence est de l'ordre de 1 % si l'anomalie est de novo, plus important si l'un des parents est porteur d'une translocation.
Trisomie 21 libre et homogène (92,5 % des cas):
cas sporadiques (de novo).
rôle de l'âge maternel (voir épidémiologie).
risque de récurrence de 1 à 2 %.
caryotype: 47,XY,+21 ou 47,XX,+21.
par non disjonction méiotique:
origine maternelle: - le division: 70 %
- 2e division: 20 %
origine paternelle:
- le division: 5 %
- 2e division: 5 %
Trisomie 21 libre en mosaïque (2,5 % des cas):
cas sporadiques.
caryotype: 46, XY / 47, XY,+21 ou 46, XX / 47, XX,+21.
accident post zygotique (mitotique).
le plus souvent exprimée par un phénotype typique, peut donner un syndrome atténué.
Trisomie 21 par translocation:
de novo ou par transmission d'une translocation parentale (équilibrée chez l'ascendant): conseil génétique surtout dans ce cas.
caryotype à 46 chromosomes: le chromosome 21 surnuméraire est transloqué le plus souvent sur un chromosome du groupe D (14, 13 ou 15) ou du groupe G (21 ou 22); exemple: 46, XY, t(14;21).
conseil génétique et risque de récurrence:
t(Dq;21q) et t(21q;22q)
maternelle: risque = 15 %
paternelle: risque = 5%
t(21q;21q): risque = 100 %:
soit --> trisomie 21
soit --> fausse couche spontanée (monosomie 21).
Autres:
trisomie 21 partielle (rareté). -->(le segment responsable de la majeure partie du syndrome est la bande 21q22.3).
associée a d'autres anomalies chromosomiques (rareté)
IV. EVOLUTION
retard statural (adulte = 1,50 m); excès pondéral (--> régime alimentaire).
La voix devient rauque.
Apparition fréquente d'une pelade.
Puberté retardée mais normale; libido faible;
Fécondité chez la femme ( --> contraceptifs).
Hypothyroïdie, Basedow (--> doser T3, T4, TSH, T3 reverse régulièrement).
Développement mental: quotient intellectuel (QI) = 50 (moyenne (et médiane)); entre 30 et 80 selon les sujets; courbe de Gauss, comme dans la population générale, mais avec une moyenne à 50 au lieu de 100; variations avec l'âge);
Insertion sociale: fonction de l'environnement familial, de l'aide qui a pu être apportée à la famille au moment du diagnostic, et de la prise en charge médico-pédagogique de l'enfant;
Rééducation psychomotrice dés 6 mois; kinésithérapie; orthophonie ensuite; école maternelle, classe intégrée si possible ensuite (classe de handicappés au sein d'une école maternelle normale: permet à la fois une meilleure intégration des trisomiques 21 et l'apprentissage de la tolérance chez les enfants normaux); institut médico pédagogique (IMP), institut médico éducatif (IME), puis - IMPro; apprentissage, activité professionnelle manuelle; les insérer dans la vie active (centre d'aide par le travail: CAT); ne doivent pas rester vivre chez leurs parents: ils y garderaient un statut infantilisant, et vieilliraient de manière encore plus accélérée du fait du vieillissement parental.
Vieillissement précoce: le comportement peut passer brusquement de l'enfant gai et sociable à l'adulte triste, inactif, inexpressif; risque de démence sénile (maladie d'Alzheimer).
V. PRONOSTIC
L'espérance de vie, faible auparavant, a été considérablement améliorée par l'utilisation de l'antibiothérapie et de la chirurgie.
le pronostic peut être grevé par:
1 - l'extrème sensibilité aux infections des trisomiques 21.
2 - les malformations, en particulier cardiaques.
3 - l'apparition d'une leucémie aigu (chez 1 % des enfants trisomiques 21, soit 20 fois plus que dans la population générale).
pronostic intellectuel: (voir évolution).
VI. TRAITEMENT
Chirurgical en cas de malformation.
Antibiothérapie des épisodes infectieux; traitement antifongique du pied d'athlète.
Prise en charge médico-pédagogique; rééducation psychomotrice; kinésithérapie; orthophonie.
Surveillance de la fonction thyroïdienne (dosages une fois par an).
Surveillance ophtalmologique (attention à la sensibilité à l'atropine), auditive.
Radio cervicale (instabilité cervicale --> risque de luxation).
Statique plantaire (chaussures, tricycle)
Dos plat (natation).
Cicatrices kéloïdes (n'opérer que si nécessaire; éviter la chirurgie esthétique).
développer et encourager la créativité artistique
Contributeurs:
Jean-Loup Huret, Pierre-Marie Sinet.