
Diagnostic anténatal
URL source du document
Document
Atlas of Genetics and Cytogenetics in Oncology and Haematology
DIAGNOSTIC PRENATAL
Introduction
I- Familles à risque
I- 1. Age maternel avancé
I- 2. Récidive des anomalies chromosomiques de nombre et de structure
I- 3. Syndrome du chromosome X fragile et retard mental
I- 4. Instabilité chromosomique
I- 5. Maladies métaboliques héréditaires
I- 6. Anomalies du tube neural
II- Echographie foetale
II- 1. L'échographie
II- 2. L'échocardiographie foetale
II- 3. Signes d'appel en échographie
II- 4. Malformations foetales dépistables par échographie au second trimestre de la grossesse
II- 4.1. Malformations du Système nerveux
II- 4.2. Malformations cardiaques
II- 4.3. Malformations thoraciques
II- 4.4. Malformations gastro-intestinales
II- 4.5. Malformations urogénitales
II- 4.6. Malformations musculo-squelettiques
II- 4.7. Autres malformations
III- Techniques de prélèvement de tissus foetaux
III- 1. L'amniocentèse
III- 2. Biopsie choriale ou choriocentèse
III- 3. Cordocentèse
III- 4. Foetoscopie
III- 5. Cellules foetales en circulation
IV- Malformations congénitales d'origine monogénique
V- Marqueurs sériques maternels
V- 1. Anomalies du tube neural
V- 2. Trisomie 21 ou syndrome de Down
VI- Hybridation in situ en fluorescence ( FISH)
VII- Perspectives d'avenir
VII- 1. Traitement in utero
VII- 2. Diagnostic pré-implantatoire
VII- 3. Le dépistage pré-conceptionnel
Introduction
Le diagnostic prénatal répond à un besoin d'identifier tôt durant la grossesse un certain nombre d'anomalies foetales ou maladies génétiques. Réalisable depuis les années soixante, le diagnostic prénatal des maladies génétiques n'est devenu pratique courante de l'évaluation des grossesses à risque qu'au cours des trois dernières décennies. Dès 1976 trois études multi-sites, réalisées en Amérique et en Europe, ont confirmé que le prélèvement de liquide amniotique au second trimestre de la grossesse, en vue d'une étude des cellules fœtales (amniocytes), était une technique fiable et peu risquée pour la mère et le fœtus.
On évalue à environ 3% le nombre de fœtus viables qui présenteraient une anomalie sévère à la naissance. L'impact du diagnostic prénatal commence à être perçu sur la fréquence des anomalies congénitales graves puisque plusieurs d'entre elles sont dépistées dès la fin du premier trimestre.
Si, de par l'anamnèse familiale ou l'histoire obstétricale, la femme est reconnue comme étant à risque de concevoir un enfant atteint d'une anomalie génétique, un diagnostic prénatal peut alors être envisagé. Ce diagnostic peut être réalisé en imagerie médicale avec ou sans prélèvement de liquide amniotique ou autre tissu d'origine fœtale selon la nature de l'anomalie recherchée. Avant de procéder à toute intervention, ou technique invasive, le sine qua non est de s'assurer qu'il y a possibilité de dépister ou d'exclure le défaut génétique qui toucherait le sujet atteint. Le diagnostic prénatal permet aux couples à risque d'envisager une grossesse puisqu'une alternative leur est maintenant offerte.
I- Familles à risque
I- 1. Age maternel avancé
Les femmes âgées de 35 ans et plus à l'accouchement ont un risque plus élevé de donner naissance à un enfant porteur d'une anomalie chromosomique par non disjonction. Ce risque accru serait dû en partie au vieillissement ovulaire. Le risque augmente avec l'âge (fig 1) et touche particulièrement la trisomie 21 (syndrome de Down) qui est la plus fréquente des anomalies autosomiques observées. Plusieurs autres, dont les trisomies 13 et 18, et les anomalies des chromosomes sexuels XXY et XXX sont également diagnostiquées plus fréquemment dans ce groupe d'âge maternel. D'un pays à l'autre les critères de sélection pour une évaluation anténatale est variable et l'amniocentèse est généralement suggérée aux femmes enceintes qui auront 35 ans ou plus à l'accouchement. Selon les disponibilités en matière de médecine préventive l'examen anténatal peut être précédé d'un test de dépistage en particulier pour la trisomie 21 à l'aide de marqueurs sériques maternels (voir marqueurs sériques). Ce test de dépistage peut permettre d'identifier dans une certaine mesure les personnes qui sont plus à risque de porter un enfant trisomique et rassurer celles qui préfèreraient ne pas subir d'amniocentèse si ce test de dépistage s'avérait négatif.
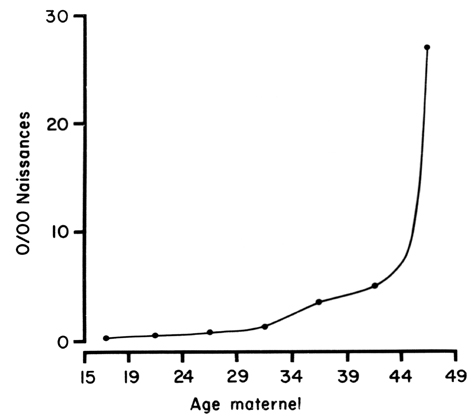
Figure 1 - Incidence de la trisomie 21 à la naissance selon l'âge maternel
I- 2. Récidive des anomalies chromosomiques de nombre et de structure
a- tout accident chromosomique (voir aussi à : Chromosomes et anomalies chromosomiques ) résultant d'une erreur de division cellulaire, ou non disjonction, entraîne un risque de récidive de plus de 1% lors d'une grossesse subséquente et ce risque peut être beaucoup plus élevé qu'attendu chez une femme âgée de 30 ans ou moins. À vingt ans le risque de trisomie 21 est d'environ 1/2000, de 1/1200 à 25 ans, 1/ 900 à 30 ans, 1/300 à 35 ans, 1/100 à 40 ans et 1/40 à 45 ans. Ce risque signifie également que l'aneuploïdie peut toucher d'autres chromosomes que le chromosome surnuméraire. Par exemple une femme qui aurait conçu un fœtus présentant une trisomie 21 pourrait, lors d'une grossesse subséquente, concevoir un enfant qui aurait une trisomie 13 ou 18, ou encore une aneuploïdie impliquant un chromosome X soit un syndrome XXX ou XXY. Il est également possible qu'une trisomie impliquant un autre autosome soit non viable et se termine en fausse couche au tout début de la grossesse.
b-translocation chromosomique familiale : un des parents est porteur d'une translocation chromosomique équilibrée et il y a risque, à des pourcentages variables, selon 1-le type de translocation, 2- l'importance des segments impliqués et 3- la ségrégation des chromosomes lors de la méiose:
de fausse couche spontanée, par déséquilibre majeur du complément chromosomique
de donner naissance à un enfant malformé ayant un complément chromosomique non équilibré
que le fœtus ait un complément chromosomique équilibré, mais tout en étant porteur sain comme un des parents
que le fœtus ait un caryotype tout à fait normal.
Dès qu'on identifie un remaniement chromosomique chez un individu la règle, dans la mesure du possible, est d'analyser le caryotype des parents et si indiqué de la fratrie, d'abord pour préciser la nature et l'origine du remaniement et par la suite prévenir les individus porteurs, mais sains, du risque de reproduction ( fig 2)
On conseille aussi aux couples une analyse de leur caryotype si ils ont une histoire personnelle ou familiale de pertes fœtales à répétition ou de naissance d'enfants avec retard mental avec ou sans dysmorphie.
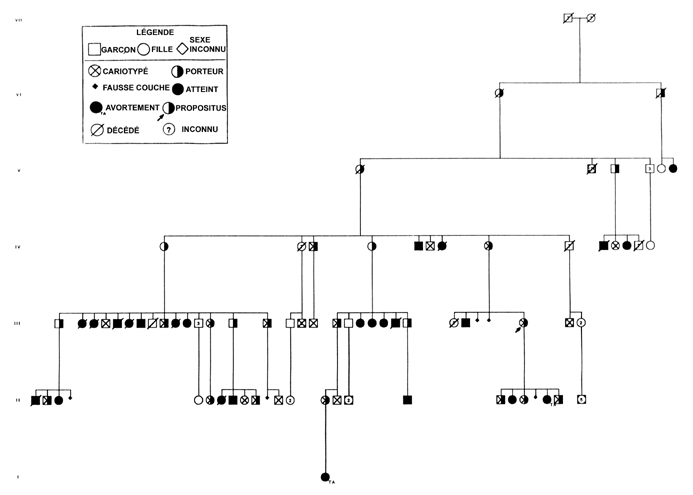
Figure 2 - Arbre généalogique partiel d'une translocation 4;18
I- 3. Syndrome du chromosome X fragile et retard mental
a- Ce syndrome (voir aussi à : Dysgonosomies et syndromes apparentés ) se caractérise chez l'enfant atteint par un faciès particulier et un retard mental de sévérité variable. Sur le plan chromosomique le signe caractéristique est une hypodensité de la chromatine dans la région Xq28 identifiable en microscopie optique : on parle alors de fragilité du chromosome X Le défaut génétique a été identifié comme étant une amplification anormale (> 60 fois) du triplet CGG au locus Xq27.3. Si l'enfant est atteint, la mère peut être porteuse ( on parle alors de pré-mutation) du syndrome elle même ayant une amplification anormale du triplet CGG (60-200). L'amplification inhibe l'expression du gène FMR-1. Les individus des deux sexes peuvent être atteints mais les mâles le sont généralement de façon plus sévère. Le syndrome du X fragile est une des causes de retard mental la plus fréquente après le syndrome de Down.
b- dans une histoire familiale de retard mental lié au X, on procédera à une étude moléculaire qui permettra d'identifier les individus atteints du syndrome X fragile et, si indiqué, un diagnostic prénatal pourra être envisagé.
I- 4. Instabilité chromosomique
Certains syndromes, called syndromes d' instabilité chromosomique se manifestent par une instabilité de la structure des chromosomes. On parle ici, à titre d'exemple, de maladie de Fanconi caractérisée par une anémie, un retard staturo pondéral, des anomalies squelettiques et du syndrome de Bloom caractérisé par une anémie, un nanisme, une hypersensibilité à la lumière. En effet il s'agit de maladies autosomiques récessives et le risque de récidive est de 25 % après la naissance d'un enfant atteint. Tous deux ont un taux élevé de cassures chromosomiques et une prédisposition au cancer. Les échanges chromosomiques sont fréquents dans la maladie de Fanconi alors que les échanges entre chromatides soeurs sont plus fréquents dans le syndrome de Bloom. En appliquant les techniques appropriées de mise en culture des cellules fœtales et de coloration des chromosomes, ces syndromes peuvent parfois se prêter à un diagnostic prénatal dans les familles à risque où les parents ont été identifiés comme étant hétérozygotes ou porteurs sains.
Toutefois la démonstration de mutations spécifiques dans ces maladies rares n'est pas toujours réalisable. Si un diagnostic prénatal est envisagé on ne peut compter sur l'étude moléculaire des cellules fœtales que si des mutations parentales ont été mises en évidence au préalable.
I- 5. Maladies métaboliques héréditaires
a- maladie métabolique diagnostiquée chez un enfant et mise en évidence d'un déficit enzymatique ou d'une mutation qui serait décelable par l'étude des cellules fœtales lors d'une grossesse subséquente.
b- maladie métabolique familiale connue: le dépistage chez le couple démontre que les deux parents sont porteurs (hétérozygotes) et ont un risque de 25 % de concevoir un enfant atteint comme dans la mucoviscidose. L'incidence élevée de cette maladie est à l'origine de programmes de dépistage d'individus porteurs d'une mutation, surtout dans les populations à risque.
I- 6. Anomalies du tube neural :
Les anomalies du tube neural sont d'origine multifactorielle et leur incidence est très variable. Elles étaient plus fréquentes dans les îles Britanniques, au Canada, en Chine et d'autres pays comme la Hongrie et atteignaient un taux de 5 pour 1000 naissances avec un risque de récidive de 5%. Aux États Unis et en France cette fréquence était plutôt de 1 pour 1000 naissances avec un faible risque de récidive.
Il a été démontré, d'abord en Grande Bretagne, que l'acide folique favorise la fermeture du tube neural et réduit le taux de récidive dans les grossesses à risque. La prise d'acide folique dès la planification d'une conception et durant les deux premiers mois pour toute grossesse, et en particulier dans les grossesses à risque, a réduit considérablement l'incidence et le risque de récidive des anomalies du tube neural dont la fermeture se complète dans les 4 premières semaines de l'embryogenèse. Il est donc fortement recommandé, lorsqu'il y a une histoire familiale d'anomalie du tube neural, de procéder à une échographie pour s'assurer de l'intégrité du développement de la voûte crânienne et du rachis. Dans les populations à risque il est très important d'encourager la prise d'acide folique dès que la personne envisage une grossesse ou abandonne tout moyen de contraception.
spina bifida aperta: lésion ouverte avec ouverture de la peau au niveau de la lésion avec ou sans extrusion de la moelle épinière;
occulta : lésion fermée .
anencéphalie: défaut de formation des os de la voûte crânienne.
Les lésions ouvertes de la moelle épinière et l'anencéphalie se manifestent par une élévation de l'alpha-foetoprotéine dans le liquide amniotique et subséquemment dans le sérum maternel. Le défaut osseux au niveau du crâne peut se manifester par une lésion circonscrite de volume variable : on parle alors d'encéphalocèle.
toute histoire de microcéphalie antérieure ou familiale doit être investiguée pour en établir l'étiologie et le mode de transmission, le défaut souvent sporadique peut également être héréditaire : autosomique dominant ou récessif. Un suivi échographique permet l'évaluation la croissance fœtale et en particulier du diamètre bi-pariétal du foetus en cours de développement, mais seules les microcéphalies sévères pourraient être décelées.
II- Échographie fœtale
II- 1. L'échographie ( fig 3) est une technique qui fait appel aux ultrasons pour examiner les tissus et les organes. L'examen se réalise dès le premier trimestre mais ce n'est qu'au second trimestre que l'on peut évaluer la morphologie fœtale de façon optimale et de préférence vers la 18ème semaine de gestation.

Figure 3 - Echographie d'un fœtus à la 16ème semaine de grossesse
II- 2. L'échocardiographie fœtale, permettant la visualisation des gros vaisseaux et des chambres cardiaques, est pratiquée de façon optimale vers la 20ème semaine.
II- 3. Signes d'appel en échographie
Les signes d'appel en échographie sont des variations, notées au cours de l'examen, qui alertent l'examinateur à la possibilité d'un développement fœtal anormal ou d'une maladie génétique. Ces signes d'appel peuvent suggérer une anomalie chromosomique telle une trisomie 21, une trisomie 13, une trisomie 18 ou encore une chondrodysplasie.
QUELQUES SIGNES D'APPEL EN ÉCHOGRAPHIE SUGGÉRANT LA PRÉSENCE D'UNE ANOMALIE FŒTALE
Anomalie cardiaque cono-troncale de la voie de chasse ventriculaire, ou du tronc commun, se manifestant par une cardiopathie complexe telle la tétralogie de Fallot ou autre malformation des vaisseaux
(syndromes de DiGeorge et vélo-cardio-facial secondaires à une délétion - del 22q11-)
Calcifications abdominales ( péritonite méconiale)
Crâne, signe du citron (spina bifida)
Défaut des coussins endocardiques ou du septum cardiaque primaire pouvant se manifester par un canal atrio-ventriculaire ( trisomie 21)
Défaut d’ossification de la voûte crânienne (anencéphalie)
Double bulle liquidienne (atrésie duodénale)
Estomac non visualisé (atrésie oesophagienne)
Flexion persistante des doigts (trisomie 18; arthrogrypose)
Fractures osseuses ( ostéogénèse imparfaite)
Hyperéchogénicité intestinale (trisomie)
Hypodensité osseuse (hypophosphatasie)
Hypoplasie faciale et fente labio-palatine ( trisomie 13, holoprosencéphalie)
Hygroma kystique ( syndrome de Turner 45,X)
Mégavessie (valve urétrale)
Persistance de kystes des plexus choroïdiens (anomalie chromosomique)
Plis du cou augmentés (trisomie 21)
Polydactylie (trisomie 13; Ellis Van-Creveld)
Pterygium colli ( syndrome de Turner; pterygium multiple)
Raccourcissement des os longs (achondroplasie)
Thorax malformé (dysplasie squelettique)
Ventricules cérébraux de volume anormal (hydrocéphalie)
II- 4. Malformations fœtales dépistables par échographie au second trimestre de la grossesse
II- 4.1 Malformations du Système nerveux
Anencéphalie
Dysplasie cranio-faciale
Encéphalocèle
Holoprosencéphalie (anomalies des ventricules cérébraux et faciales associées)
Hydrocéphalie
Kyste de la fosse postérieure
Microcéphalie
Myéloméningocèle
Porencéphalie (formation kystique intra-cérébrale)
Rachischisis (défaut étendu de la fusion vertébrale)
Spina-bifida
II- 4.2 Malformations cardiaques
Anomalie vasculaire
Défaut septal
Défaut valvulaire
Épanchement péricardique
Hyperplasie ventriculaire
Hypoplasie ventriculaire
Situs inversus
Troubles du rythme cardiaque
II- 4.3 Malformations thoraciques
Atrésie congénitale de l'œsophage
Épanchement pleural
Hernie diaphragmatique
Kystes intrathoraciques
II- 4.4 Malformations gastro-intestinales
Absence de muscles abdominaux
Ascite
Atrésie intestinale
Hernie diaphragmatique
Kyste du mésentère
Laparoschisis ( éviscération para-ombilicale du tube digestif)
Lymphangiome kystique
Omphalocèle (hernie à l'ombilic de viscères abdominaux)
Tumeur du cordon (choriohémangiome)
II- 4.5 Malformations urogénitales
Agénésie rénale
Hydronéphrose
Hydrouretère
Reins polykystiques
Tératome sacro-coccygien
Valve urétrale
II- 4.6 Malformations musculo-squelettiques
Absence de mouvements des membres
Arthrogrypose
Défaut de minéralisation osseuse
Défaut de réduction (amélie ou absence de membre)
Fractures
Dysplasies osseuses
Pied(s) bot(s)
Ptérygium colli,
Ptérygium multiple,
II- 4.7 Autres malformations
Brides amniotiques
Kystes
Monstre acardiaque
Jumeaux siamois
Tératomes
Tumeurs
III- Techniques de prélèvement de tissus fœtaux
III- 1. L'amniocentèse
L'amniocentèse ou ponction de liquide amniotique (fig 4) est dite précoce si elle est réalisée vers la 12ème semaine de gestation. Bon nombre de cliniques favorisent l'amniocentèse entre la 14ème et 16ème semaine. Des études randomisées ont démontré une incidence élevée de perte de liquide amniotique si le prélèvement est réalisé avant la 12ème semaine et un risque accru de malformations squelettiques en particulier de pieds bots secondaires à l'oligoamnios. On prélève de façon stérile de 10 à 30 ml de liquide selon l'âge de la grossesse(fig 5). Des cellules fœtales d'origine de la voie digestive haute et du système urinaire, de la peau et des membranes baignent dans ce liquide et sont récupérées par centrifugation du spécimen.
Elles sont mises en culture pour une période de 5 à 10 jours en présence de sérum fœtal et d'un milieu nutritif favorisant la croissance cellulaire. La multiplication cellulaire est alors suffisante pour permettre la préparation des lames sur lesquelles on retrouve des cellules au stade de métaphase. La numération des chromosomes au microscope et l'étude de leur structure est alors complétée. Un traitement du matériel cellulaire, lors de la préparation des lames, met en évidence des zones plus ou moins claires correspondant au marquage chromosomique. Ces zones sont le reflet du ratio variable de nucléotides A-T ;G-C sur les chromatides.
Au besoin des analyses enzymatiques ou moléculaires peuvent être prescrites sur ces échantillons cellulaires.
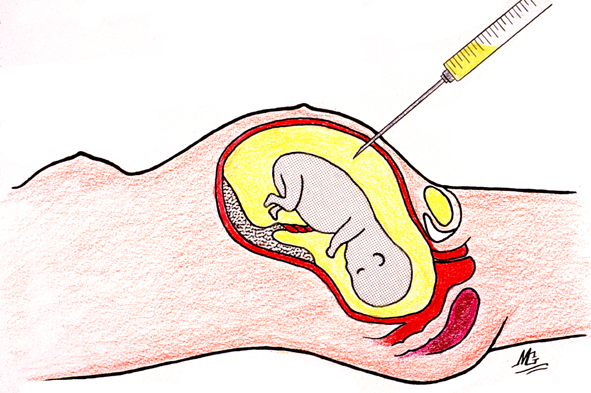
Figure 4 - Schéma illustrant la procédure d'amniocentèse
III- 2. Biopsie choriale ou choriocentèse
Le prélèvement de villosités choriales par voie vaginale (fig 6) donne accès à des cellules fœtales dont plusieurs sont en voie de division et peuvent être analysées dans les heures qui suivent le prélèvement. Un risque plus élevé de pertes fœtales et de contamination du spécimen par des cellules maternelles a convaincu plusieurs cliniciens d'abandonner ce type de prélèvement réalisé avant la 12ème semaine. Quelques rapports ont fait état du risque d'anomalie de réduction des membres si la biopsie est faite vers la fin du premier trimestre. Dans certaines circonstances, pour lesquelles le risque de récidive est élevé comme par exemple dans les maladies métaboliques héréditaires ou encore si un des parents est porteur d'une translocation chromosomique équilibrée, cette approche permet alors l'obtention d'un résultat en quelques jours vers la 11ème ou 12 ème semaine de grossesse.

Figure 5 - Schéma illustrant la procédure de biopsie choriale
III- 3. Cordocentèse
On peut prélever du sang du cordon sous guidage échographique. Cette voie d'accès permet une analyse à court terme du complément chromosomique ou métabolique vers la fin du second trimestre lorsqu'il y a urgence de préciser un diagnostic : résultats non concluants des premières analyses, menace d'accouchement L'analyse cytogénétique rapide, à partir des lymphocytes, permet la confirmation ou l'exclusion d'une anomalie chromosomique suspectée à l'étude des amniocytes. Cette approche peut également être utile pour préciser une mosaïque comme par exemple la trisomie 20 en mosaïque dont le pronostic est généralement favorable ou identifier un marqueur chromosomique qui serait confiné aux annexes. Cette voie a été favorisée pour l'étude de déficits immunitaires sévères entre autres pour la mesure de l'adénosine déaminase et l'examen des cellules T.
III- 4. Foetoscopie
La foetoscopie consiste à visualiser le fœtus, de préférence vers la fin du second trimestre, en introduisant dans l'utérus, par voie transabdominale et sous guidage échographique, un tube muni de fibres optiques permettant de pratiquer une biopsie ou d'autres manipulations chirurgicales. Pour des raisons évidentes de sécurité cette technique invasive n'est que rarement utilisée en clinique.
III- 5. Cellules fœtales en circulation
La présence de cellules fœtales en circulation dans le sang maternel pourrait nous renseigner sur le complément chromosomique ou le génotype du fœtus. Des recherches en ce sens sont en cours depuis plusieurs années et n'ont à date donné que peu de résultats sur l'efficacité de cette technique non invasive de diagnostic prénatal. Les embûches sont multiples soit la rareté des cellules nuclées, leur isolation, leur identification et l'analyse génétique. L'apport du FISH et autres techniques d'identification chromosomique et le PCR pour l'analyse moléculaire ont récemment encouragé les chercheurs à ne pas abandonner cette voie qui pourrait révolutionner l'approche du diagnostic prénatal des maladies génétiques.
IV- Malformations congénitales d'origine monogénique
Plusieurs malformations congénitales ont une composante héréditaire et leur mode de transmission peut être autosomique dominant, récessif ou encore lié au chromosome X. Ces syndromes pourront être diagnostiqués au second trimestre de la grossesse par échographie si ils s'accompagnent d'un vice de développement ( par exemple l'achondroplasie) ou encore par la recherche d'une mutation. Par contre dans les histoires antérieures d'anomalie fœtale d'origine indéterminée, mais visible à l'échographie, la mère pourra être rassurée par un examen échographique normal si elle devient enceinte à nouveau. Entre autres cette mesure s'applique pour le laparoschisis qui est l'extériorisation para ombilicale des anses intestinales et parfois du foie et de l'omphalocèle isolé ou hernie des viscères abdominaux pour lesquels le risque de récidive est minime. L'omphalocèle peut se retrouver avec d'autres syndromes dont la trisomie 13.
V- Marqueurs sériques maternels
Les marqueurs sériques sont des protéines normales en circulation chez la mère et dont la mesure permet de dépister un certain nombre de pathologies fœtales au début de la grossesse. Ainsi l'efficacité du dépistage est augmentée si les deux approches, échographie fœtale et marqueurs sériques, sont complétées simultanément.
V- 1. Anomalies du tube neural
En premier lieu l'alpha-1 foeto protéine (AFP) constitue 20% des protéines fœtales en circulation et varie suivant l'âge de la grossesse au moment où elle est mesurée (fig 7). Une augmentation de l'AFP dans le liquide amniotique se reflète par transudation dans le sérum maternel et signe la présence d'une lésion ouverte du tube neural chez le fœtus. Une valeur élevée dans le liquide amniotique peut, dans certains cas, signaler la présence d'une lésion cutanée non évidente à l'échogaphie ou d'une souffrance fœtale.

Figure 6 - Valeurs d'AFP dans le liquide amniotique au second trimestre
V- 2. Trisomie 21 ou syndrome de Down
l'AFP peut également nous mettre sur la piste d'une trisomie 21 si la valeur sérique maternelle est abaissée selon l'âge de la mère vers la 16ème semaine de grossesse.
l'ensemble de trois marqueurs, désigné sous le nom de triple test, comprend la gonadotrophine chorionique humaine (hCG), l'oestriol ( uE3) et l'AFP. La mesure de ces trois protéines, en fonction du nombre de semaines de gestation, peut donner un risque approximatif, que le fœtus ait une trisomie 21. La fiabilité de ce test est de l'ordre de 60 % ou plus selon les critères d'évaluation et le taux de faux positifs retenu.
un dépistage plus précoce, basé sur la mesure de la clarté nucale
de l'hCG et d'une protéine placentaire la PAPP-A, est offert à la fin du premier trimestre et jusqu'à la 13ème semaine. La clarté nucale correspond à l'épaisseur de la nuque. Une mesure supérieure à 3 mm est considérée comme suspecte à la 12ème semaine de grossesse. Ce dépistage est fiable dans environ 80 % des cas de trisomie 21 et peut mettre sur la piste d'autres pathologies fœtales. Un résultat anormal sera précisé par une étude chromosomique des cellules fœtales.
VI- Hybridation in situ en fluorescence ( FISH)
L'hybridation in situ en fluorescence consiste en l'utilisation de sondes moléculaires marquées en fluorescence et correspondant à un gène ou une séquence d'ADN donnant un signal visible au microscope à la lampe UV à un endroit précis d'un chromosome. Le FISH peut s'appliquer dans un premier temps aux cellules fœtales au stade d'interphase dès le prélèvement du liquide amniotique et confirmer la présence d'un complément euploïde ou aneuploïde entre autres pour les chromosomes X,Y, 13,15,18, 21.Cette technique s'applique à l'étude de marqueurs chromosomiques qui, si marqués, peuvent être décelés dans les amniocytes. En d'autres circonstances le FISH peut être utilisé pour identifier l'origine de segments supplémentaires ou confirmer la perte de séquences précises ou délétion sur un chromosome donné.
VII- Perspectives d'avenir
VII- 1. Traitement in utero
Le traitement in utero se limite surtout aux ponctions liquidiennes ( mégavessie, épanchements pleuraux, kystes…..) D'autres approches chirurgicales sont envisagées et ont fait l'objet d'essais soit la pose d'un cathéter intra-vésical, la correction de certaines malformations cardiaques, la réduction de l'hydrocéphalie, l'ablation de tumeurs et autres manipulations. La mise en évidence et la caractérisation de malformations de la paroi abdominale ( ex : omphalocèle) permet de prévoir une planification du mode d'accouchement et des interventions immédiates chez le nouveau-né et éviter les complications systémiques de telles anomalies.
Sur le plan médical l'arythmie cardiaque fœtale peut être traitée par une approche médicamenteuse maternelle. Finalement une diète restrictive en phénylalanine, amorcée dès la planification d'une grossesse chez la mère phénylcétonurique, préviendra les complications fœtales dont une microcéphalie et un retard mental sévère chez l'enfant..
Le dépistage prénatal de certaines maladies métaboliques comme la galactosémie ou la leucinose permet de planifier, si le fœtus est atteint, une diète restrictive appropriée à la naissance afin de prévenir les complications précoces pour l'enfant secondaires aux métabolites anormaux.
VII- 2. Diagnostic pré-implantatoire
Le diagnostic pré-implantatoire consiste en l'analyse d'une cellule prélevée d'un œuf fécondé qui se trouve para exemple au stade de 8 cellules. Le diagnostic pré-implantatoire a été introduit dans les techniques de procréation assistée en 1989 mais on considère qu'il s'agit d'une manœuvre qui est toujours au stade de recherche et développement et dont on ne connaît pas encore la fiabilité et la sécurité bien qu'à date les quelques douzaines de nouveau-nés, issus de cette pratique, semblent avoir un développement normal. Seulement quelques laboratoires en Europe et en Amérique ont les facilités et le savoir faire pour développer cette technique et l'offrir comme moyen de diagnostic.
à date cette technique a été utilisée dans plus d'une centaine de grossesses à risque dont surtout la mucoviscidose et moins fréquemment la dystrophie musculaire de Duchenne, l'hémophilie A, la thalassémie alpha et beta, l'atrophie bulbaire, le syndrome de Lesch Nyhan, l'incontinence pigmentaire, la mucoviscidose, la maladie de Huntington, la dystrophie myotonique.
pour les maladies liées au chromosome X on s'assure d'abord qu'il s'agit d'un fœtus de sexe mâle avant de poursuivre les analyses indiquées qui sont généralement de nature moléculaire.
VII- 3. Le dépistage pré-conceptionnel
Le dépistage pré-conceptionnel est une avenue nouvelle dans le domaine de la prévention. Il implique la recherche d'anomalies au niveau des gamètes et de façon plus spécifique au niveau de l'ovule. Des publications récentes font état de diagnostic pré-conceptionnel en utilisant le premier corps polaire d'ovules en voie de maturation : les corps polaires étant l'image du complément chromosomique et du génotype de l'ovule qui pourrait être utilisé pour une fécondation in vitro. Dans un exemple cité dans la littérature une mère étant porteuse d'un gène dominant pour une forme sévère de maladie d'Alzheimer on sélectionna chez elle un ovule normal et indemne de mutation ce qui lui aurait permis de rendre à terme un enfant normal. La voie est donc ouverte aux interventions diagnostiques, qui font appel à l'étude des gamètes, mais des dilemmes éthiques découleront de toute tentative de manipulation des cellules germinales.
Contributeurs:
Louis Dallaire