La Mucoviscidose et le gène CFTR
Atlas of Genetics and Cytogenetics in Oncology and Haematology
La MUCOVISCIDOSE et le GÈNE CFTR
I- Historique
II- Épidémiologie
III- Clinique
IV- Diagnostic biologique
V- Le gène CFTR et ses mutations
V-1. introduction
V-2. La mutation F508
V-3. Spectre des mutations du gène CFTR
V-4. Corrélations génotype-phénotype
VI- La protéine CFTR et ses fonctions
VI-1. Structure de la protéine
VI-2. Fonctions de la protéine
VI-2.1. Fonction canal Cl-
VI-2.2. La protéine CFTR, une protéine multi-fonctionnelle
VI-3. Corrélations des mutations du gène CFTR avec la fonction canal Cl-
VI-3.1. Classe 1 : mutations altèrant la production de la protéine.
VI-3.2. Classe 2 : mutations perturbant le processus de maturation cellulaire de la protéine.
VI-3.3. Classe 3 : mutations perturbant la régulation du canal Cl-.
VI-3. 4. Classe 4 : mutations altèrant la conduction du canal Cl-.
VI-3. 5. Classe 5 : mutations altérant la stabilité de l'ARNm CFTR. VI-3. 6. Classe 6 : mutations altérant la stabilité de la protéine mature.
*
I- Historique
La description de la mucoviscidose en tant qu'affection autonome date de la fin des années 30: l'association "fibrose kystique congénitale du pancréas et bronchectasies" est identifiée en 1936.
En 1953, est mis en évidence un excès de chlorure de sodium dans la sueur des enfants atteints. Cette découverte conduira, peu après, à la mise au point du "test de la sueur", seul test de diagnostic positif de la maladie actuellement disponible.
Dans les années 80, l'anomalie du transport de sels fut précisée: défaut de perméabilité aux ions chlorure (Cl-) affectant les cellules épithéliales des glandes sudoripares, et au niveau de l'épithélium respiratoire.
Mais la nature biochimique du défaut à l'origine de la maladie est restée longtemps inconnue, ce qui a contraint les chercheurs à employer les outils de la "génétique inverse" (ou clonage positionnel). En 1985, le locus CF fut localisé sur le bras long du chromosome 7 grâce à la découverte d'une liaison avec un site polymorphe exploré par une sonde anonyme située sur ce chromosome.
En 1989, le gène CFTR , impliqué dans la mucoviscidose, a été isolé. Ce gène est localisé en 7q31 et contient 27 exons. La protéine est composée de 1480 acides aminés.
II- Épidémiologie
C' est la plus fréquente des maladies autosomiques récessives graves dans les populations d'origine européenne, touchant 1/2500 nouveau-nés (= q2), ce qui permet d'évaluer à un sur 25 (= 2pq) les individus transmetteurs (hétérozygotes). Cependant la fréquence de l'affection varie selon l'origine géographique et ethnique des patients.
III- Clinique
Présentation clinique: très polymorphe tant entre les différentes familles qu'au sein d'une même famille. Chez la plupart des patients le diagnostic est posé avant l'adolescence, mais quelques uns restent asymptomatiques jusqu'à l'âge adulte. Cliniquement et biologiquement, il n'est pas possible de distinguer les hétérozygotes des sujets non porteurs d'allèle muté (allèle CF).
Circonstances de découverte de la maladie: variables selon l'âge; révélé dès la naissance par un iléus méconial (occlusion intestinale due à un méconium anormalement épais) chez 10% des nouveaux-nés atteints. Plus tard la symptomatologie peut associer des infections respiratoires à répétition avec des signes en rapport avec une malabsorption digestive.
Atteinte respiratoire: prédominante; liée à une obstruction des bronchioles par un mucus épais et visqueux propice à la croissance des micro-organismes. Ceci explique en partie les infections répétées à germes opportunistes.
Atteinte digestive avec insuffisance pancréatique exocrine (défaut de production des enzymes): notée chez 85% des patients, conséquence de l'obstruction des canaux excréteurs et à l'origine d'une maldigestion lipidoprotidique et d'une malabsorption. La fonction pancréatique exocrine, déficiente (PI ou pancreatic insufficiency) ou conservée (PS ou pancreatic sufficiency), permet d'apprécier la gravité du phénotype des patients: phénotype grave dans le premier cas, modéré dans le second.
Anomalies des glandes sudoripares: conduisent à un excès de chlorure de sodium dans la sueur, cette perte de sel pouvant être responsable de déshydratation aiguë en cas d'exposition à la chaleur.
Autres organes qui peuvent être touchés: en particulier l'appareil génital et le foie.
98% des hommes atteints sont stériles en raison d'une azoospermie obstructive (agénésie des canaux déférents), tandis que 80% des femmes atteintes sont fertiles.
L'atteinte hépatique (hépatomégalie dans 30% des cas, une insuffisance hépatique dans 9% des cas) est liée à l'obstruction des voies biliaires intra-hépatiques ou extra-hépatiques par compression au niveau du pancréas. Dans 2 à 5% des cas ces lésions conduisent à une cirrhose biliaire dans des délais variables.
Pronostic:
En l'absence de traitement, la médiane de survie est de 3 à 5 ans.
Il n'existe toujours pas de traitement efficace de la mucoviscidose mais la précocité du diagnostic ainsi que l'efficacité de l'antibiothérapie et de la kinésithérapie permettent d'augmenter l'espérance de vie des malades, qui est actuellement de 25 à 30 ans en moyenne.
Les traitements actuels sont symptomatiques : kinésithérapie respiratoire, antibiothérapie, nébulisation de bronchodilatateurs et mucolytiques, administration d'inhibiteurs de protéases pour les manifestations pulmonaires et apport d'enzymes pancréatiques de substitution et de vitamines pour pallier l'insuffisance pancréatique.
Une transplantation coeur-poumon voire coeur-poumons-foie n'a eu lieu que dans les atteintes très évoluées.
La thérapie génique rencontre de nombreux obstacles. D'autres stratégies prometteuses sont actuellement à l'étude.
IV- Diagnostic biologique
Le diagnostic positif de la mucoviscidose repose sur le test de la sueur. C'est encore aujourd'hui l'examen le plus fiable pour dépister la maladie. Les techniques utilisées actuellement sont simples mais la quantité de sueur recueillie qui peut être insuffisante chez le nourrisson de moins de deux mois, le risque d'erreur pouvant alors atteindre 30%.
Un taux sudoral de chlorures inférieur à 40 mmoles/l est généralement considéré comme normal.
Pour des taux compris entre 40 et 60 mmoles/l, l'interprétation est douteuse et il faut recommencer le test.
Le diagnostic est positif lorsque des taux supérieurs à 60 mmoles/l sont retrouvés sur plusieurs examens successifs.
Une autre technique consiste à mesurer la différence de potentiel transépithélial (DDPTE) qui existe entre la peau et la muqueuse nasale. Cette valeur est significativement augmentée en cas de mucoviscidose. Son intérêt concerne essentiellement trois types de situations:
1) le diagnostic précoce chez le nouveau-né présentant une pathologie digestive suspecte, alors que le test de la sueur est encore difficile à réaliser;
2) les diagnostics douteux où sont associés des signes cliniques évocateurs avec des tests de la sueur intermédiaires ou négatifs;
3) le suivi évolutif des patients puisqu'il existe une corrélation entre la gravité de l'atteinte respiratoire mesurée par l'étude du VEMS (volume expiré maximal par seconde) et les mesures de DDPTE. La méthode est simple et peu coûteuse.
V- Le gène CFTR et ses mutations
V-1. introduction
Le gène CFTR contient 27 exons s'étendant sur 250 kb du chromosome 7, en 7q31, et code un ARNm de 6,5 kb.
V-2. La mutation F508
La mutation la plus fréquente, une délétion de trois nucléotides aboutissant à l'élimination de la phénylalanine en position 508 (F508) rend compte de 70% des allèles CF.
Il existe une grande hétérogénéité de répartition de cette anomalie, suivant un gradient nord-ouest/sud-est en Europe, avec, par exemple, 88% de F508 au Danemark, 81% en Bretagne et 50% en Italie.
Cette fréquence élevée dans les populations nord-européennes suggère l'existence possible 1- d'un important effet fondateur (un seul événement mutationnel survenu dans le passé) et 2- d'un avantage sélectif des hétérozygotes. Les hétérozygotes seraient protégés contre la déperdition hydrique et saline au cours des diarrhées dues à des toxines bactériennes. La protéine DF508 diminuerait par ailleurs l'entrée du germe pathogène dans l'épithélium intestinal assurant une protection vis à vis de l'infection.
V-3. Spectre des mutations du gène CFTR
· Plus de 900 mutations ont été décrites ( http://www.genet.sickkids.on.ca/cftr/ ), dont quatre, hors DF508, représentent plus de 2% des cas. Toutes les autres mutations sont rares, voire détectées seulement dans une famille. La fréquence de certaines mutations varie énormément d'un groupe géographique à l'autre (ex:la mutation W1282X touche 48% des allèles CF chez les Juifs ashkénazes et seulement 2% des allèles CF totaux).
· Types de mutations: La majorité des défauts moléculaires du gène CFTR sont des mutations ponctuelles réparties comme suit: 42% de mutations faux-sens, 24% de microinsertions et microdélétions entrainant un décalage de la phase de lecture, 16% de mutations non-sens, 16% de mutations d'épissage et 2% de délétions d'un acide aminé. Quelques grandes délétions sont également rapportées.
· Transcrits délétés chez les sujets normaux: une des particularités du gène CFTR est l'existence de transcrits délétés d'un ou de plusieurs exons chez des sujets normaux. Ces transcrits sont dus à des anomalies conduisant à un épissage alternatif, dont le transcrit délété de l'exon 9 (9-). La présence ou l'absence de cet exon est corrélé avec un "polymorphisme" (5T, 7T ou 9T) de séquence de l'intron 8 situé près du site accepteur d'épissage. Si 7T ou 9T permet d'assurer un épissage normal à 90%, 5T ne permet de produire que 10 à 40% d'ARNm normal fonctionnel
V-4. Corrélations génotype-phénotype
· Environ la moitié des patients atteints de la mucoviscidose sont homozygotes pour la mutation DF508. A l'état homozygote, DF508 est associée à la forme classique de la maladie avec une augmentation des électrolytes dans la sueur, une insuffisance pancréatique et une atteinte des poumons le plus souvent sévère.
· Etant donné la fréquence élevée de DF508 (66 %), 40 % des patients sont hétérozygotes composites avec DF508 sur un allèle et une autre mutation du gène CFTR sur l'autre chromosome.
· D'une manière générale, la fonction pulmonaire, l'âge de début de la maladie et le taux de chlore sudoral sont peu corrélés à un génotype particulier.
· D'autre part , hormis pour l'état de la fonction pancréatique, la variabilité au sein d'une même fratrie laisse prévoir que le génotype seul du gène <CITE&>CFTR ne peut expliquer totalement le phénotype.
· Seule la mutation A455E (patients
hétérozygotes composites DF508/A455E) a été associée à une atteinte pulmonaire
modérée.
- Une ou deux mutations faux-sens permettrait une fonction pancréatique
conservée (PS ou pancreatic sufficiency), alors que deux allèles avec
mutations d'épissage, non-sens, ou décalant la phase de lecture entrainent une
insuffisance pancréatique (PI ou pancreatic insufficiency). Les patients
ayant une mutation PI sur un allèle et une mutation PS sur l'autre allèle ont
un phénotype PS; donc, avec une mutation PS permet une activité CFTR suffisante
au niveau pancréatique.
· L'effet d'une mutation peut être modulé par une deuxième mutation héritée en cis sur le même allèle.
· Le polymorphisme de la séquence polypyrimidique de l'intron 8 module la pénétrance de certaines mutations.
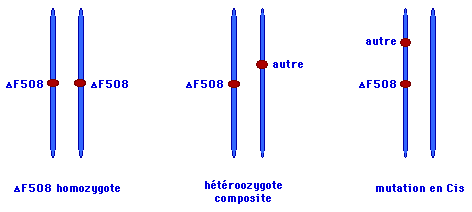
Mutations. Editor.
VI- La protéine CFTR et ses fonctions
VI-1. Structure de la protéine
· La protéine est composée de 1480 acides aminés.
· Elle contient deux motifs répétés constitués chacun d'un domaine hydrophobe transmembranaire (TMD) contenant six hélices a et d'une importante région hydrophile contenant des séquences susceptibles de lier l'ATP (NBF ou Nucleotide Binding fold). Ces deux motifs sont reliés par un domaine cytoplasmique (domaine R) codé par l'exon 13, contenant de nombreux résidus chargés et la majorité des sites potentiels de phosphorylation (substrats probables des protéines kinases A et/ou C)
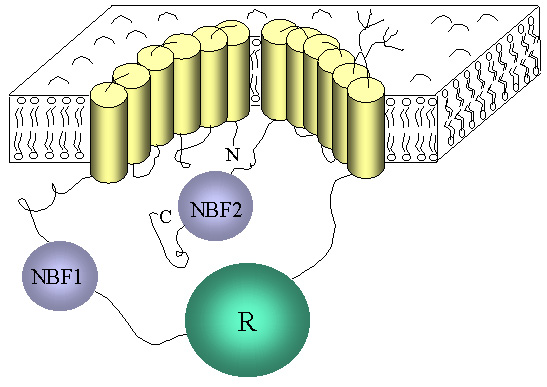
Figure 1: Structure prédictive de la protéine CFTR, díaprès Riordan et al. 1989 - Pascale Fanen.
· Une homologie de séquence primaire existe avec les membres d'une famille de protéines membranaires, la famille des transporteurs ABC (ATP-binding cassette) qui transportent activement des substrats au travers des membranes cellulaires, l'hydrolyse de l'ATP fournissant de l'énergie à ce transport.
VI-2. Fonctions de la protéine
VI-2.1. Fonction canal Cl-
La première hypothèse postulait que la protéine CFTR était un canal Cl-.
Cette hypothèse était compatible avec le défaut de perméabilité aux ions Cl-
de la membrane apicale des épithéliums CF. L'autre hypothèse proposait que la
protéine CFTR n'était pas un canal ionique mais qu'il agissait sur la
régulation des canaux Cl- soit en s'y associant, soit en
transportant, hors ou dans la cellule, un facteur régulateur des canaux Cl-.
Les données expérimentales ont procuré des preuves irréfutables de la fonction
canal Cl- de la protéine CFTR.
VI-2.2. La protéine CFTR, une protéine multi-fonctionnelle
Les découvreurs du gène CFTR ont baptisé son produit
"régulateur transmembranaire d'une conductance". En effet, la
protéine CFTR régule d'autres canaux, le canal Cl- à rectification
sortante, le canal Na+ épithélial, et au moins deux canaux K+appartenant
à la famille de canaux K+ à rectification entrante.
D'autres fonctions, indépendantes de la régulation des canaux, ont été
décrites: transport d'ATP, modulation des phénomènes d'exocytose/endocytose,
régulation du pH des organelles intracellulaires....
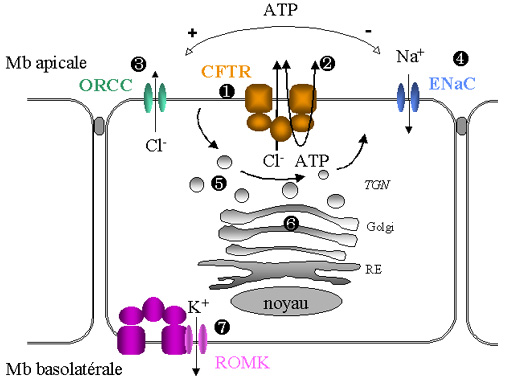
Figure 3: CFTR, une protéine multifonctionnelle, díaprès Schwiebert et al. 1999 - Pascale Fanen.
VI-3. Corrélations des mutations du gène CFTR avec la
fonction canal Cl-
Les anomalies moléculaires ont des conséquences variables sur la protéine CFTR
et sa fonction. Une classification de ces anomalies par rapport à la fonction
canal Cl- aété proposée (Figure 2):
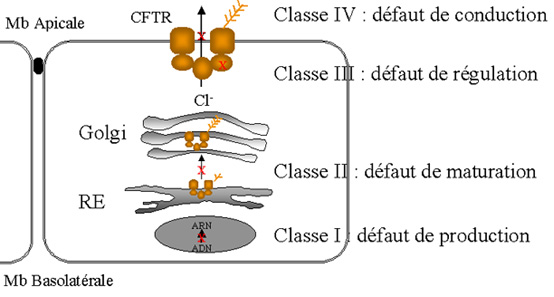
Figure 2: Classification des mutations du gène CFTR, díaprès Welsh & Smith, 1993 - Pascale Fanen.
VI-3.1. Classe 1 : mutations altèrant la production de la protéine. Ces
mutations résultent en une absence totale ou partielle de la protéine. Cette
classe inclut les mutations non-sens et celles qui produisent un codon stop
prématuré (anomalies d'épissage et mutations décalant la phase de lecture).
Dans certains cas, l'ARNm muté est instable et ne produit pas de protéine. Dans
les autres cas, la protéine anormale produite sera probablement instable et
rapidement dégradée. C'est ce qui se produit quand la protéine est tronquée ou
contient des séquences aberrantes (anomalies d'épissage ou de décalage de la
phase de lecture). Sur le plan fonctionnel, ces mutants devraient conduire à
une perte de la conductance au Cl- du canal CFTR dans les
épithéliums atteints.
VI-3.2. Classe 2 : mutations perturbant le processus de maturation
cellulaire de la protéine. De nombreuses mutations altèrent la maturation de la
protéine et son ciblage vers la membrane plasmique. Ainsi, la protéine est soit
absente, soit présente en quantité réduite dans la membrane apicale. Les
mutations de cette classe représentent la majorité des allèles CF (ex: DF508).
VI-3.3. Classe 3 : mutations perturbant la régulation du canal Cl-.
Ces mutations sont le plus souvent situées dans les domaines de liaison à l'ATP
(NBF).
VI-3. 4. Classe 4 : mutations altèrant la conduction du canal Cl-.
Certains segments des domaines transmembranaires participent à la formation du
pore ionique. Les mutations faux-sens situées dans ces régions produisent une
protéine correctement positionnée qui présente une activité canal Cl--AMPc
dépendante. Mais les caractéristiques de ces canaux sont différentes de celles
du canal CFTR endogène avec une diminution du flux d'ions et une sélectivité
modifiée.
VI-3. 5. Classe 5 : mutations altérant la stabilité de l'ARNm CFTR.
VI-3. 6. Classe 6 : mutations altérant la stabilité de la protéine
mature.
Contributeurs: Pascale Fanen